
Contextualizing the Heritability of Schizophrenia

This is a follow-up to my last post:

I want to focus on how we can use the failure of Genome Wide Association Studies (GWAS) to produce strong genetic associations to revisit the traditional understanding of schizophrenia as a highly heritable condition. Doesn't the heritability of schizophrenia, which is estimated to be ~80%, constitute strong evidence of the genetic causation of schizophrenia, that schizophrenia is “80% genetic”? Not quite. I think schizophrenia is 80% genetic only in the technical sense of heritability, but not in any other sense.
Heritability is a population-level metric and doesn’t apply to individuals.
Heritability is a technical term in genetics and refers to a population-level phenomenon. It refers to the degree of variation in a phenotypic trait in a population that is due to genetic variation between individuals in that population. This is a pretty standard qualification offered in the genetics literature. E.g., Robette, et al. (2022): “As we have seen, heritability is not an individual characteristic but a population measure. It does not tell anything on the genetic determinism of the trait under study.”
We cannot, for example, use schizophrenia’s 80% heritability at the population level to say, “For a randomly selected person with schizophrenia in this population, 80% of their etiology is genetic in nature.” The heritability statistic applied to an individual has no meaning. (See Moore and Shenk, 2017, for a discussion of this.)
Heritability doesn’t tell us about causal processes operating in an individual.
Since heritability concerns itself with variation across individuals in a population, it doesn’t say anything directly about causal processes that occur within any individual person. In particular, if a person develops schizophrenia, pointing to heritability doesn’t tell us that the development of schizophrenia in this individual is caused in any meaningful way by genetic factors. Relying on heritability to say that “a certain trait’s appearance in an individual is caused by genetic factors” is recognized as a misunderstanding of heritability. A statistical investigation focused on variation is not necessarily informative about the causal mechanisms that give rise to any individual instance of a phenomenon (Moore and Shenk, 2017).
Here is Robette, et al. (2022):
“there is often a confusion between the contribution of genetic factors to the phenotype and their contribution to the phenotype variability. Heritability says nothing about the causes, the mechanisms at the origin of differences between populations, nor about the etiology of diseases. As Lewontin (1974) reminded us, there is a crucial distinction between the analysis of variance and the analysis of causes. A strong heritability does not mean that the main factors involved in the trait are genetic factors.”
Heritability estimates rely on important assumptions that can be violated.
Heritability estimates assume, for example, that genetic influences on trait development can be separated from environmental influences in such a manner that we can talk about the relative contributions of each. However, to the extent that genetic influence is the result of a dynamic interaction with the environment, this assumption is an oversimplification. I will sidestep the issue of whether this invalidates the notion of heritability or not (and assume that it doesn’t), but it nonetheless highlights that heritability estimates could be misleading if this dynamic relationship between genes and environment is ignored.
Estimates of heritability using twin studies also rely on the assumption that environments encountered by identical twins are no more similar than environments encountered by fraternal twins. This is not entirely true either. For example, “It starts in the womb: while fraternal twin embryos are always connected to their mother via two unique placentas, identical twins most often (but not always) share a single placenta. This means that, beginning soon after conception, identical twins typically have more similar access to nutrients, oxygen, and other factors than do fraternal twins” (Moore and Shenk, 2017) Again, this may not invalidate the heritability estimates entirely, but it can certainly inflate the estimates inaccurately. This assumption is particularly important for schizophrenia since intrauterine events such as infections during pregnancy, maternal inflammation, maternal nutritional deficiencies, obstetric emergencies, maternal use of cannabis, etc. are believed to be relevant to schizophrenia risk.
80% heritability is consistent with a much smaller risk at an individual level
This is evident from twin studies, especially twin studies in which twins are selected using population-based samples. Here is the table from Torrey 2024:
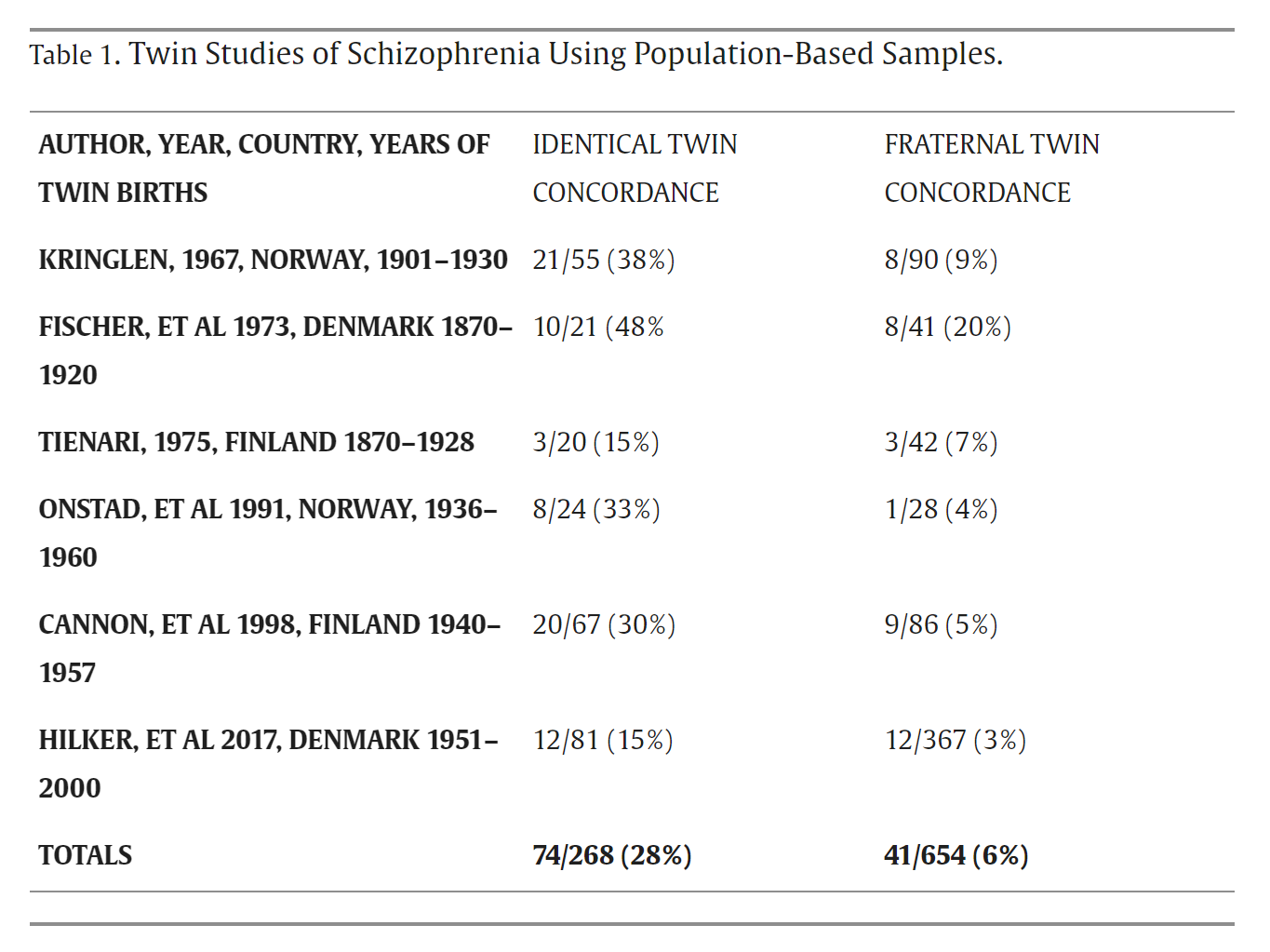
On average, across these studies, if one identical twin has schizophrenia, the likelihood that both twins have schizophrenia is 28%. In the largest of these studies, Hilker et al. 2017, the concordance is 15%. For fraternal twins, the concordance is 6%. Importantly, these figures are consistent with 80% heritability at the population level. The heritability estimate in Hilker et al. was 79%, and yet, in the same sample, there was only a 15% chance that both twins would have schizophrenia if at least one twin had been diagnosed. This is yet more evidence that “schizophrenia is 80% genetic” is kind of meaningless when applied to individuals.
Schizophrenia is highly polygenic, yes, but…
As we discussed in the last post, schizophrenia is influenced by hundreds or possibly thousands of genes, each conferring a tiny risk (with some rare variants having a larger effect). Most complex disorders and behavioral traits are polygenic in this sense. However, it is not clear to me that, from the polygenic nature of schizophrenia, we can make the inference that in order for someone to develop schizophrenia, they must have hundreds or thousands of schizophrenia-associated genes present in their genome. I may be wrong on this (and I am happy to be shown wrong, if I am), but I don’t see how we can move from “schizophrenia is heritable and polygenic” to “most patients who develop schizophrenia, develop it due to a genetic vulnerability.” That may turn out to be true, but it’s not evident simply from heritability or polygenicity itself. As noted in the prior post, the genes associated with schizophrenia are not specific to schizophrenia. We can think of them as conferring a nonspecific vulnerability to psychopathology rather than conferring a risk for schizophrenia per se. This is not troubling as such, but it is something that must be made explicit. It does raise the question of whether we can call them genes “for schizophrenia” in some meaningful sense. In a classic 2005 paper in the American Journal of Psychiatry, Ken Kendler discusses:
“A central phrase in the new “GeneTalk” is “X is a gene for Y,” in which X is a particular gene on the human genome and Y is a complex human disorder or trait. This article begins by sketching the historical origins of this phrase and the concept of the gene-phenotype relationship that underlies it. Five criteria are then proposed to evaluate the appropriateness of the “X is a gene for Y” concept: 1) strength of association, 2) specificity of relationship, 3) noncontingency of effect, 4) causal proximity of X to Y, and 5) the degree to which X is the appropriate level of explanation for Y. Evidence from psychiatric genetics is then reviewed that address each of these criteria. The concept of “a gene for…” is best understood as deriving from preformationist developmental theory in which genes—like preformationist anlagen—“code for” traits in a simple, direct, and powerful way. However, the genetic contribution to psychiatric disorders fails to meet any of the five criteria for the concept of “X is a gene for Y.” The impact of individual genes on risk for psychiatric illness is small, often nonspecific, and embedded in complex causal pathways. The phrase “a gene for…” and the preformationist concept of gene action that underlies it are inappropriate for psychiatric disorders.”
It may very well be the case that schizophrenia is heritable and polygenic, and yet there are no “genes for” schizophrenia.
What are our standards for “genetic etiology”?
Given that GWAS has failed to reveal common genes that confer more than a tiny risk, are we simply relying on the high heritability to say that schizophrenia is a “genetic disorder”? That may be fine, if we are clear on what we are doing. But I am inclined to think that our standards for privileging genetics in this manner should be higher.
Take the case of epilepsy. Epilepsy is a recurrent abnormal, excessive, and synchronized electrical discharge in the brain. It has a heritability of 70-88%, at par with schizophrenia. However, epilepsy is not broadly characterized as a genetic disorder. The 2017 ILAE Classification of Epilepsies recognizes six etiologic categories: genetic, structural, metabolic, immune, infectious, and unknown.
“The concept of a genetic epilepsy is that it results directly from a known or presumed genetic mutation in which seizures are a core symptom of the disorder.” (Epilepsia)
The inference that a genetic etiology is present can be made based on the family history of an autosomal dominant disorder, for example. In cases where genes are not yet identified, there can be situations where "a molecular basis may have been identified and may implicate a single gene or copy number variant of major effect.”
Clearly, only a very small subset of people with schizophrenia have a genetic etiology in this sense. 22q11.2 deletion syndrome would be one contender. Other CNVs and rare variants with larger effects could be candidates as well, etc.
Aggregate genetic influences matter. They are particularly relevant when it comes to biological relatives with schizophrenia. Yet, for us to move beyond the abstraction of heritability, genetic research has to deliver something tangible in the form of genes that matter. Maybe one day we’ll have a schizophrenia polygenic score of a thousand genes that will make aggregate genetic influences tangible. I don’t deny the possibility, but it is also not guaranteed. Furthermore, a critical re-evaluation of the notion of heritability (as above) makes any inference from an “analysis of variance” to an “analysis of causes” convoluted and uncertain. As discussed in the previous post, it is proving to be more productive to use GWAS signals to look for relevant mechanistic pathways instead of looking for genes with substantial effects. Once we recognize that a pathway exists, we can also recognize that the pathway can be disrupted in a wide variety of ways, many of which won’t involve genes at all.
“The end result is a hypothesized cascade that goes from complement activation, synapse elimination, and cortical thinning to cognitive impairments and aberrant information processing. It is a cascade that points away from genetic causes towards pathways that are disrupted in different individuals through a combination of a variety of different factors, which may include infection, inflammation, cannabis use, childhood trauma, adversity in later life, etc.”
I don’t deny that genetic influences exist, that schizophrenia has high heritability, or that schizophrenia is highly polygenic, but I am uncertain what it all means for the actual causal role played by genes in the pathophysiology of schizophrenia. Heritability is “biologically vacuous” (Matthews & Turkheimer, 2022), and I think we would be better off if more of us hesitated to assert that schizophrenia is a “genetic disorder” based predominantly on heritability estimates.
Excellent discussion, Awais--thank you! I would like to shift figure and ground for a moment, and note the importance of epigenetic factors. As Dr. Joe Coyle et al point out in a seminal paper on schizophrenia (I thank Dr. Manuel Mota for this reference):
"...adults who experienced abuse as children have altered epigenetic states and decreased expression of the glucocorticoid receptor gene NR3C1 in the hippocampus as compared with control subjects without a history of childhood abuse (61, 62). Epigenetic mechanisms are especially important in the complex pathophysiology of psychiatric disorders, including schizophrenia, where they mediate the emergence of molecular pathologies in specific populations of cells in specific brain regions stemming from nonspecific genetic and environmental risk factors. Fetal viral infections, famine during pregnancy, and complicated birth (63) as well as childhood abuse and neglect (64) are established environmental risk factors for schizophrenia."
And finally, as Bleuler pointed out in 1911, we are almost certainly dealing with a group of related disease processes (hence, Bleuler's term, "schizophrenias"). Accordingly, it may be more useful to inquire about the genetic and epigenetic factors (not to mention the psychosocial ones!) in the etiology and pathogenesis of the schizophrenias (plural).
Regards,
Ron
Ronald W. Pies, MD
1. Coyle J. Fifty Years of Research on Schizophrenia: The Ascendance of the Glutamatergic
Synapse. https://ajp.psychiatryonline.org/doi/10.1176/appi.ajp.2020.20101481
Journal articles and books I have read by some of these authors and others, including Surviving Schizophrenia by E. Fuller Torrey, The Developing Genome by David S. Moore, and Innate by Kevin J. Mitchell suggests to me that many authors begin with preconceived beliefs about the etiology of schizophrenia and then seek evidence in support of those beliefs that is often unrepresentative of what really is a broad lack of consensus in academia. For example, Torrey cites what are tiny studies to question genetics before arguing for toxoplasmosis as a cause for schizophrenia, a position that has been questioned by other researchers who find less compelling results. Moore does a nice job of explaining heritability and epigenetics. But he also writes extensively about problematic beliefs in genetic determinism that may be rarer than he suggests among geneticists and biochemists: While teaching genetic determinism may be a problem among faculty members who either have not kept current in their field or are trying to simplify a topic for undergraduate students, I found when reading Emery and Rimoin's Principles and Practices of Medical Genetics and Genomics that not a single one of the many geneticists and biochemists who authored chapters of that book believe in genetic determinism--in fact, most of them explicitly reject the idea while explaining that genes and the proteins they encode do not exist in a vacuum but interact with their environments. On the other hand, authors of recent studies of adverse childhood experiences (ACEs) find those studies may often overemphasize the role of environment: One published as "Population vs individual prediction of poor health from results of adverse childhood experiences screening" (2021) in the Journal of the American Medical Association (JAMA) and another titled "Poor individual risk classification from adverse childhood experiences screening" (2022) in the American Journal of Preventive Medicine concluded that ACEs are only useful on a population level and not at the individual patient level where a careful assessment of a patient's entire family history is necessary. So wrong-headed thinking about genetic determinism may also apply to weakly supported thinking about infectious agents, trauma, and other purported causes of schizophrenia with each of those also identifying only a tiny amount of risk associated with (i.e., not causative of) schizophrenia. For these reasons, and because most researchers know a lot more than I do about these topics, I take no position.